
| 產品編號 | bsm-54072R |
| 英文名稱 | PMP22 Recombinant Rabbit mAb |
| 中文名稱 | 外周髓鞘蛋白-22重組兔單抗 |
| 別 名 | GAS3; CMT1A; CMT1E; DSS; GAS-3; Growth Arrest Specific 3; Growth arrest-specific protein 3; HMSNIA; HNPP; MGC20769; Peripheral Myelin Protein 22; PMP-22; PMP22; PMP22_HUMAN; Sp110; Trembler. |
| 研究領域 | 免疫學 神經生物學 糖蛋白 |
| 抗體來源 | Rabbit |
| 克隆類型 | Recombinant |
| 克 隆 號 | 1A11 |
| 交叉反應 | Human,Rat |
| 產品應用 | WB=1:500-2000
not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 理論分子量 | 22 kDa |
| 檢測分子量 | |
| 細胞定位 | 細胞膜 |
| 性 狀 | Liquid |
| 濃 度 | 1mg/ml |
| 免 疫 原 | Recombinant human PMP22 protein(Full length) |
| 亞 型 | IgG |
| 純化方法 | affinity purified by Protein A |
| 緩 沖 液 | 0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| 保存條件 | Store at -20℃ for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antib |
| 注意事項 | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| PubMed | PubMed |
| 產品介紹 |
PMP22 is a 22 kDa glycoprotein expressed in the compact myelin of the peripheral nervous system. In the peripheral nervous system, PMP 22 is produced by myelinating Schwann cells and is coexpressed with the genes for myelin basic protein (MBP) during nerve development and regeneration. Alterations in the level of this protein cause several genetic human diseases. If the protein is duplicated, patients develop Charcot Marie Tooth disease. If one copy of the gene is deleted, they suffer from the inherited tendency to pressure palsies. Function: Might be involved in growth regulation, and in myelinization in the peripheral nervous system. Subcellular Location: Cell membrane; Multi-pass membrane protein. DISEASE: Defects in PMP22 are the cause of Charcot-Marie-Tooth disease type 1A (CMT1A) [MIM:118220]; also known as hereditary motor and sensory neuropathy IA. CMT1A is a form of Charcot-Marie-Tooth disease, the most common inherited disorder of the peripheral nervous system. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathy or CMT1, and primary peripheral axonal neuropathy or CMT2. Neuropathies of the CMT1 group are characterized by severely reduced nerve conduction velocities (less than 38 m/sec), segmental demyelination and remyelination with onion bulb formations on nerve biopsy, slowly progressive distal muscle atrophy and weakness, absent deep tendon reflexes, and hollow feet. CMT1A inheritance is autosomal dominant. Defects in PMP22 are a cause of Dejerine-Sottas syndrome (DSS) [MIM:145900]; also known as Dejerine-Sottas neuropathy (DSN) or hereditary motor and sensory neuropathy III (HMSN3). DSS is a severe degenerating neuropathy of the demyelinating Charcot-Marie-Tooth disease category, with onset by age 2 years. DSS is characterized by motor and sensory neuropathy with very slow nerve conduction velocities, increased cerebrospinal fluid protein concentrations, hypertrophic nerve changes, delayed age of walking as well as areflexia. There are both autosomal dominant and autosomal recessive forms of Dejerine-Sottas syndrome. Defects in PMP22 are a cause of hereditary neuropathy with liability to pressure palsies (HNPP) [MIM:162500]; an autosomal dominant disorder characterized by transient episodes of decreased perception or peripheral nerve palsies after slight traction, compression or minor traumas. Defects in PMP22 are the cause of Charcot-Marie-Tooth disease type 1E (CMT1E) [MIM:118300]; also known as Charcot-Marie-Tooth disease and deafness autosomal dominant. CMT1E is an autosomal dominant form of Charcot-Marie-Tooth disease characterized by the association of sensorineural hearing loss with peripheral demyelinating neuropathy. Defects in PMP22 may be a cause of inflammatory demyelinating polyneuropathy (IDP) [MIM:139393]. IDP is a putative autoimmune disorder presenting in an acute (AIDP) or chronic form (CIDP). The acute form is also known as Guillain-Barre syndrome. Similarity: Belongs to the PMP-22/EMP/MP20 family. SWISS: Q01453 Gene ID: 5376 Database links: Entrez Gene: 5376 Human Omim: 601097 Human SwissProt: Q01453 Human Unigene: 372031 Human Unigene: 1476 Rat 神經生物學相關蛋白(Neurobiology) 外周髓鞘蛋白22(PMP22)是一種糖蛋白,在外周神經系統中的致密肌纖維素中表達。它由髓鞘雪旺氏細胞產生,并在神經發育和再生過程中與MBP和Po蛋白共同表達。該蛋白表達水平變化可引起幾種人類遺傳性疾病,如果該蛋白增多,則病人會發生Charcot-Marie-Tooth疾病,如果缺失,則易患壓力麻痹的遺傳傾向。 |
| 產品圖片 |
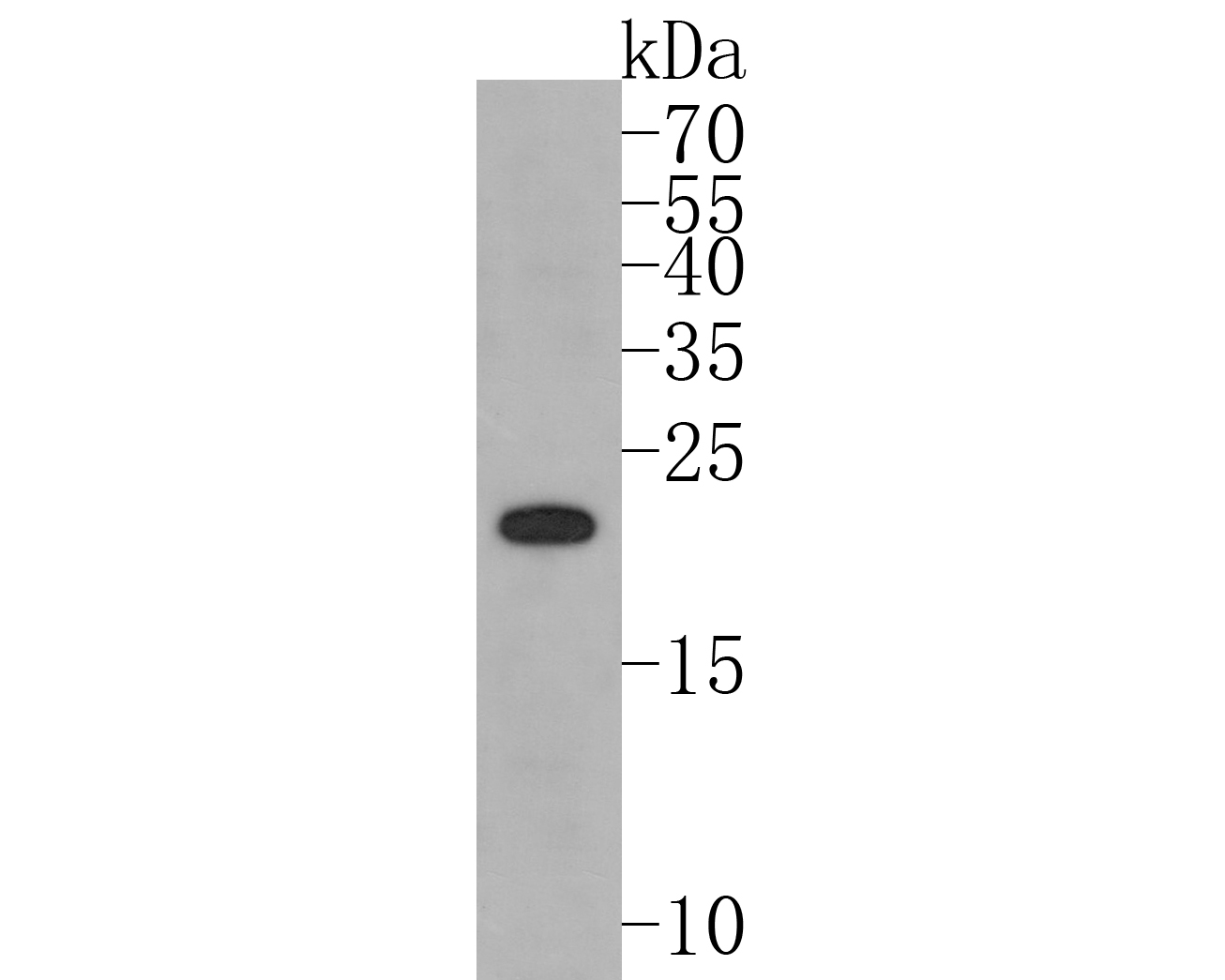
Western blot analysis of PMP22 on PC-12 cell lysates. Proteins were transferred to a PVDF membrane and blocked with 5% BSA in PBS for 1 hour at room temperature. The primary antibody (bsm-54072R, 1/500) was used in 5% BSA at room temperature for 2 hours. Goat Anti-Rabbit IgG - HRP Secondary Antibody (HA1001) at 1:5,000 dilution was used for 1 hour at room temperature.
|
| 1、抗體溶解方法 | |
| 2、抗體修復方式 | |
| 3、常用試劑的配制 | |
| 4、免疫組化操作步驟 | |
| 5、免疫組化問題解答 | |
| 6、Western Blotting 操作步驟 | |
| 7、Western Blotting 問題解答 | |
| 8、關于肽鏈的設計 | |
| 9、多肽的溶解與保存 | |
| 10、酶標抗體效價測定程序 | |




